



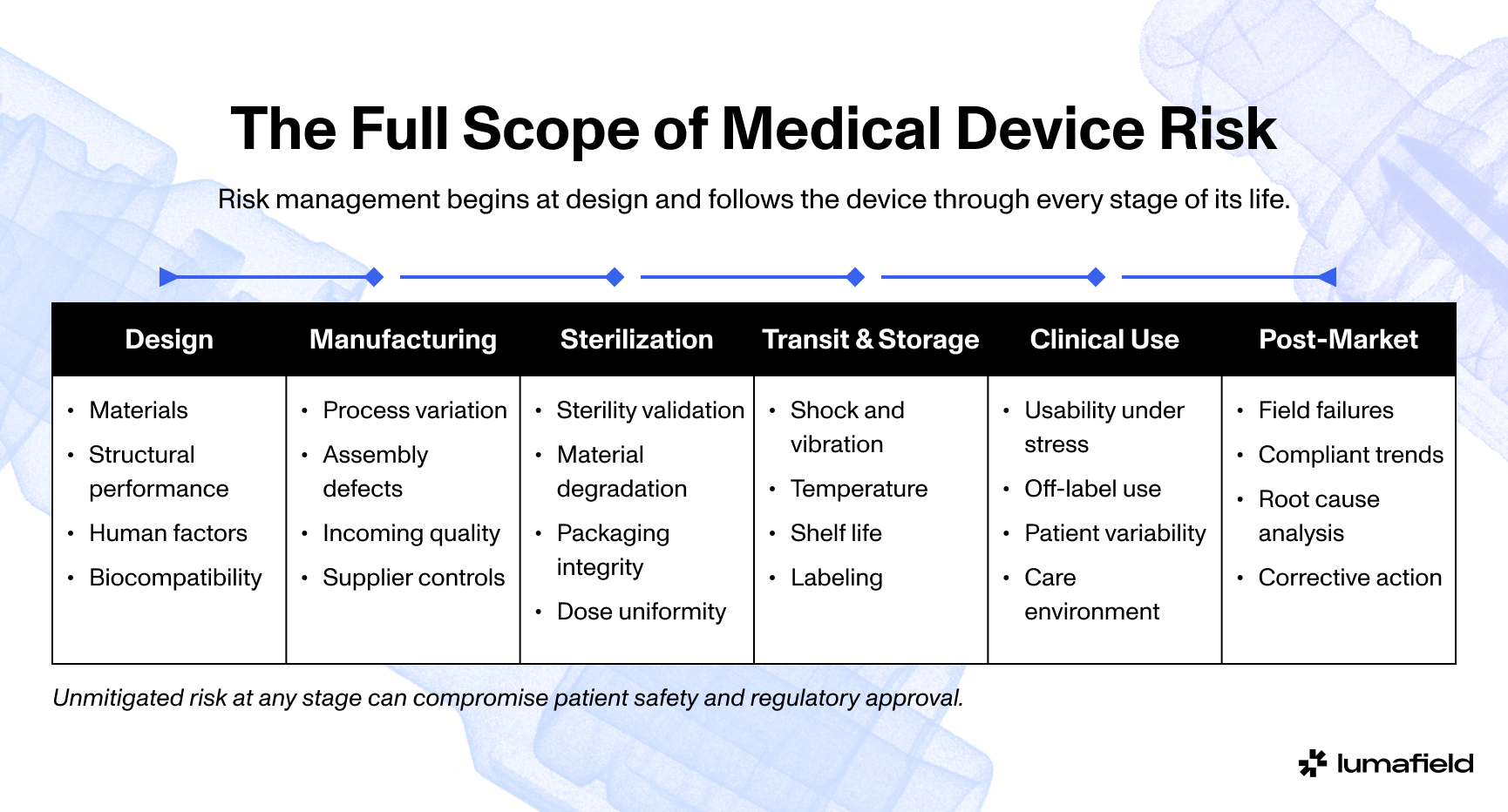
The FDA's February 2026 transition to the Quality Management System Regulation, or QMSR, has captured the attention of the entire medical device industry, and rightly so. For many teams, particularly those already operating under ISO 13485, the practical implications are less dramatic than the headlines suggest. For others, especially companies that have only ever sold into the US market, the shift requires real preparation. Understanding what changes and where the gaps tend to appear is the most useful place to start.
What QMSR and ISO 13485 change in practice
For companies already certified to ISO 13485, the move to QMSR is not a major disruption. If you are selling globally or into Europe, you have been integrating risk management through the product lifecycle already, and in some respects the alignment is a relief: one standard to track rather than two that occasionally pull in different directions.
The companies with more work ahead are those that have operated exclusively under the FDA's previous Quality System Regulation without ISO certification. For them, the most important immediate step is a thorough gap analysis: reviewing their current quality management system and standard operating procedures against the new requirements, and identifying where their documentation and processes fall short. The area that tends to surface the most gaps is risk management, specifically the expectation that risk thinking is woven through the entire product lifecycle, not treated as a discrete activity at specific checkpoints.
Risk-based thinking across the product lifecycle
Risk, in the regulatory sense, is a broader concept than most people assume when they first encounter it. It is not just about whether a device might break. It encompasses everything that can affect product quality from the moment manufacturing is complete through the full lifecycle of the device in the field: transit, sterilization, storage, handling by clinical staff, interaction with the patient. Any variable that is not understood and controlled contributes to overall product risk.
In practice, manufacturers plan and manage this through a structured process. There is typically an overarching risk management file, with subsidiary plans and files covering areas such as design, production, and usability. Each of those feeds into a cumulative risk profile. The goal is to understand the variables and outside forces that may impact patient safety and product quality, and the process ensures that no high risks go unmitigated, and that the aggregate of lower-level risks does not reach a point where the product should not be released. As risk cannot be eliminated entirely, this understanding and mitigation is critical to successful and safe product launches.
The process follows a straightforward logic: what is the initial risk, what is the response to it, and what is the residual risk after that response? Regulators are not looking for perfection. They are looking for rigor: evidence that teams have thought carefully about risk at every stage, documented their reasoning, and put controls in place that are proportionate to what they found.
The audit gaps most teams miss
For companies preparing for their first audit under QMSR, the gap that appears most often is a disconnect between how risk management is understood internally and what regulators expect to see demonstrated, and it rarely shows up as a missing document.
The best teams treat risk management as more than just a compliance activity, more than checking a box as part of a design history file. The regulatory expectation aligns with that thinking: risk management should be embedded in the way the business operates rather than being bolted onto it. That means having documented risk assessments at the appropriate stages of the product lifecycle, ensuring that the assumptions driving those assessments are interrogated rather than carried forward uncritically, and maintaining the traceability to show how risk was tracked and addressed as the product evolved.
ISO 14971, the standard for QMS risk management, is a useful reference point here. Checking your risk management file against the standard, and ensuring your documentation reflects the right assessments at the right lifecycle stages, is a concrete and practical way to close the most common gaps before an audit.
The evidence that regulators increasingly expect
Risk analysis is only as credible as the evidence supporting it. Two categories of evidence have become increasingly central to how quality teams build that credibility.
The first is failure mode characterization. Testing products to failure, understanding how and why they fail, and documenting those failure modes rigorously is foundational. This is not new, but the expectation of depth and traceability has increased. Knowing your failure modes well also creates a significant advantage later: if a device is returned from the field, you are starting from a position of knowledge rather than having to reconstruct the root cause from scratch.
The second is real-time statistical process control (SPC), brought as close to the source of manufacturing as possible. No manufacturing process produces identical results every time. There are too many variables. But robust insights and trending data on your inspection equipment, maintained continuously rather than sampled at checkpoints, give you the ability to see problems forming before they result in nonconforming product. That is the direction quality teams should be moving: from reactive inspection to predictive process monitoring.
More data, or better data, generated with the same level of investment, translates directly into greater confidence in your risk analysis. That confidence is what regulators are ultimately looking for.
Verifying that what you build matches what you designed
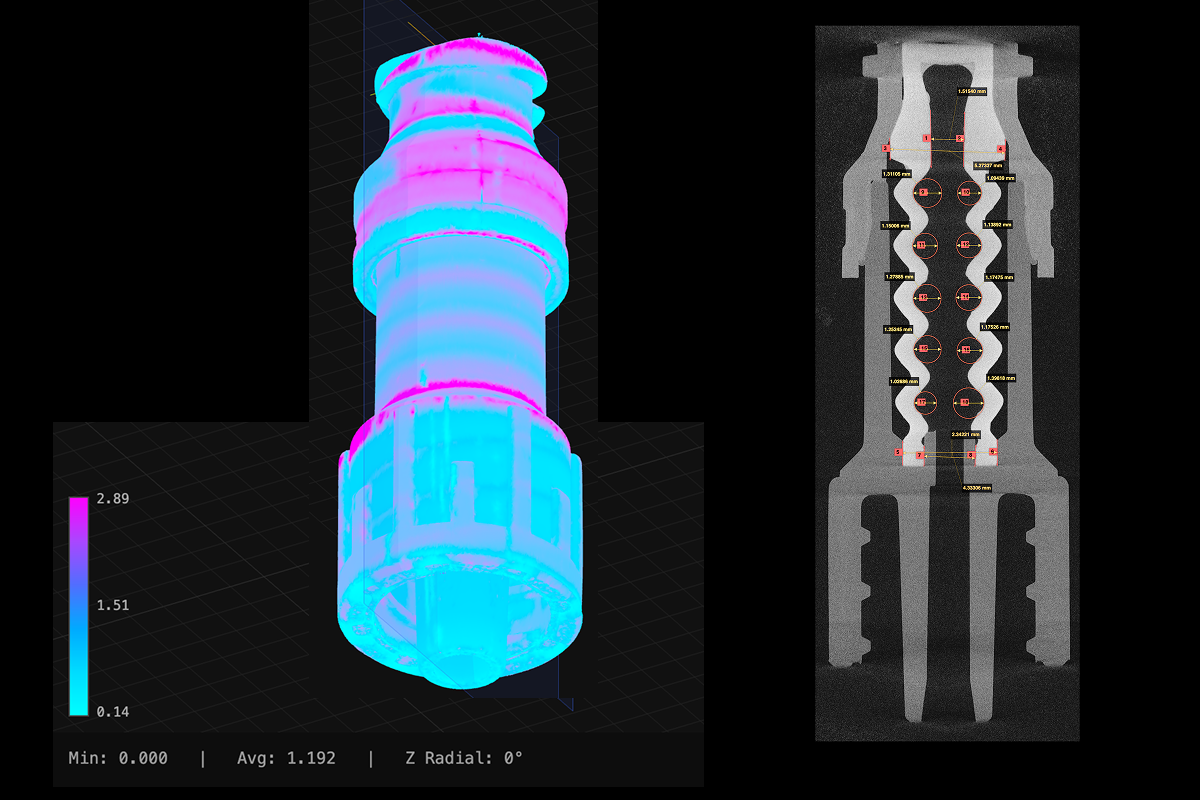
One of the more persistent challenges in medical device manufacturing is the gap between design intent and production reality. A device that performs well in development can behave differently at scale, and the variation is often subtle enough that checkpoint-based inspection misses it.
The most effective approach is identifying the features that are truly critical to quality, understanding what deviations in those features actually mean for product performance, and then building inspection and verification around them rather than around a general sampling protocol. This allows quality teams to generate richer, more targeted data, reduce the number of parts destroyed in functional testing, and maintain a clearer line of sight between manufacturing variation and product risk.
When something goes wrong in the field
Field failures, when they occur, require rapid and credible root cause analysis. The most valuable data for that process typically comes from returned devices. If a device can be retrieved and analyzed in the lab, a team that has thoroughly documented its known failure modes is in a fundamentally different position than one that has not. Rather than re-investigating failure modes from first principles, they can compare what they are seeing against a library of documented failures and understand quickly whether this is a known mode, a variation of one, or something new.
This is also where machine learning is beginning to have a concrete and meaningful impact. Training a model on documented failure modes from the design process allows post-market surveillance teams to work with better data, flag anomalies faster, and identify edge cases that may not have been anticipated during development. That kind of capability, connecting design-phase knowledge to field performance data, is one of the more practical and immediate applications of AI in medical device quality today.
Building a quality system that holds up under scrutiny
For earlier-stage companies and startups, the QMSR requirements can feel like a significant compliance burden. The more useful framing is to think of them as a design problem: how do you build risk-based thinking into the way your team works rather than layering it on top of existing processes as a documentation exercise?
The most important principle is thoughtful interpretation of the regulation for your specific context. There is no single correct implementation. What matters is that your approach is coherent, that it genuinely reflects how you manage risk, and that it produces the traceability to demonstrate that. Build it into your business processes, not just your QMS. Establish checkpoints throughout the product development lifecycle where the assumptions driving your risk analysis are revisited and stress-tested. And keep the end goal in view: a product that surgeons and clinicians can rely on, with a post-market record that reflects the rigor of what went into building it.
The next five to ten years
The next five to ten years in medical device development will be shaped significantly by the integration of new tools into quality and risk management workflows. Machine learning, AI-assisted analysis, and digital twins are beginning to make it possible to understand products faster and with more precision than was previously achievable. The value of those tools, in a regulatory context, is that they can accelerate the process of understanding what level of risk control is needed to produce a device that is both safe and reliably manufacturable.
The underlying expectation from regulators is not changing: demonstrate that you understand your product, that you understand the risks, and that your controls are proportionate and traceable. What is changing is the sophistication of the tools available to meet that expectation. The companies that learn to use them well will be better positioned not just for audits, but for the harder and more important goal of putting better devices into clinical use.